





欧盟新规申报难度超FDA,强制公告机构门槛提高!
2017年5月5日,欧洲联盟(EU)发布了新版医疗器械法规MDR(EU2017/745),以替代旧版医疗器械指令MDD(93/42/EEC)和有源植入医疗器械指令AIMDD(90/385/EEC),同年5月26日生效,过渡期三年。过渡期内,医疗器械厂商可以自主选择以旧指令MDD或新法规MDR申请CE证书;过渡期结束将强制执行新版MDR法规。

欧盟医疗器械法规历史进程节点(动脉网制图)
然而,原计划于2020年5月26日强制执行的新版MDR法规,由于受到疫情等相关因素影响,被宣告推迟一年,直到今年5月26日才正式强制执行。
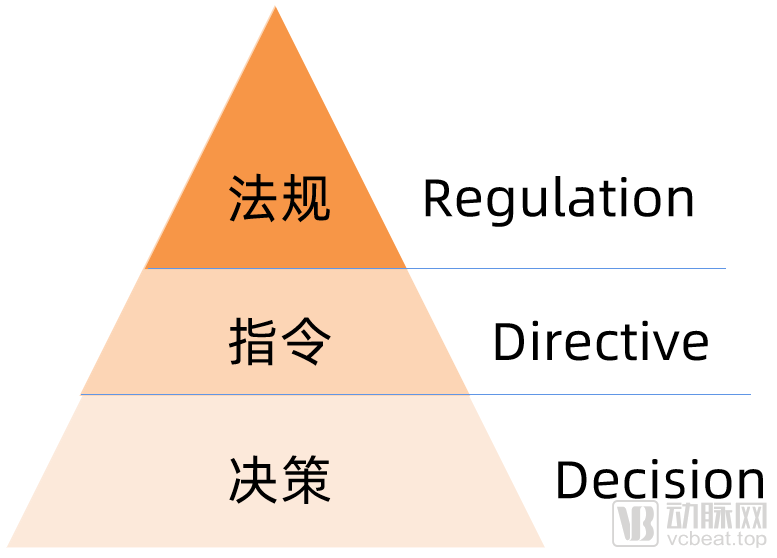
欧盟法规的金字塔体系(约束力由高到低)
从Directive(指令)到Regulation(法规),欧盟提高了对医疗器械的约束力,发布立即在欧盟成员国生效并成为有约束力的法律,此次的Regulation无需像Directive那样需要经过成员国转化成当地法律法规再去落地实施。
值得一提的是,在欧盟医疗器械分类当中,将医疗器械划分为医疗器械(MD)和体外诊断器械(IVD)两大类,目前受到新规MDR管辖的仅限于MedicalDevices,体外诊断器械的相应新规IVDR(EU2017/746)的执行时间为2022年5月26日,也就意味着IVD器械厂商还有一年的时间可以缓冲和适应新规变化,并做好应对准备工作。
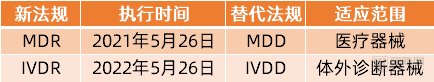
欧盟的两类医疗器械法规及执行时间
对于其他医疗器械(MD)厂商来说,从2021年5月26日开始,如果要申请欧盟认证则必须遵从新规MDR执行。相对于旧法令MDD,欧盟新法规MDR发生了哪些变化?面对这些变化,国产医药器械厂商在申请CE认证时需要注意的点有哪些?对于之前已经申请CE认证的厂商,未来如果继续持有CE证书需要做好哪些应对工作?
欧盟新规申报难度超FDA,强制公告机构(NB)门槛提高
在过去,欧盟CE认证的难度较中国NMPA及美国FDA更低,背后的原因则是欧盟医疗器械旧法规的约束力松弛,对医疗器械研发商报批要求普遍较低。同时也导致了部分仅获得CE认证的医疗器械在欧盟地区落地后也常出现医疗事故,所以这些产品未来进入中国及美国市场时,仍然需要面临更长期、更严格的报批流程。
在新规执行之前,一些低风险医疗器械产品研发厂商在申请CE认证时,可以通过自我声明的方式进行申请,但这种申请的方式监管并不严格、缺乏约束力。所以在2020年业内还出现了一则曝光事件,中国深圳某医疗器械公司产品通过自我声明方式出口欧盟,最终发现90%产品不合格。
如果产品没办法精准管控,那么CE认证的“含水量”便较高。据业内人士透露,针对欧盟这种情形,英国、德国甚至还出现过“小名单”——如果你的产品只在欧盟拿了认证,那么该产品在英德申报时还是需要经历严格的临床试验,以确保产品的安全性、有效性。
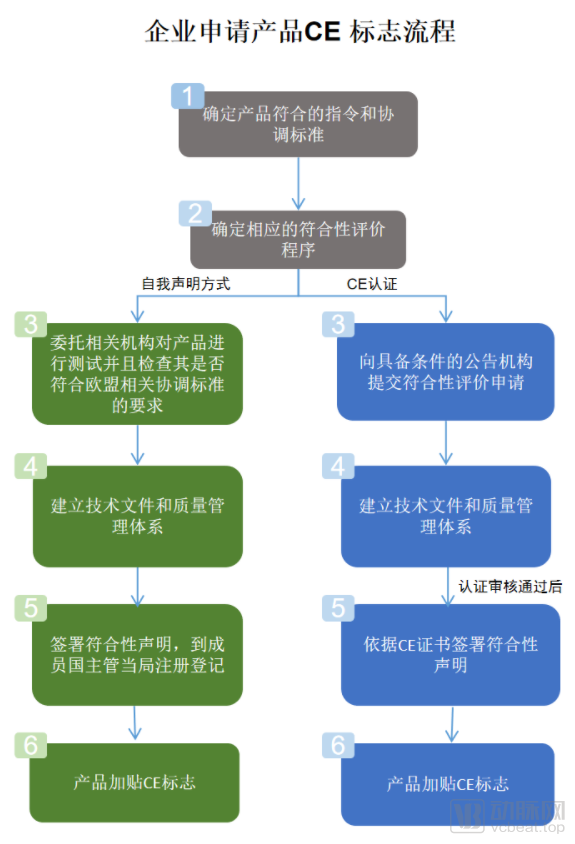
欧盟医疗器械新旧法规申报流程变化(截图自关务小二&中国国际贸易促进委员会官网)
但在欧盟MDR新规执行之后,即便是一些电商平台上进行销售的医用产品也需要通过严格的授权公告机构(Notified Body,NB/欧代)进行申报。与此同时,欧盟对NB机构的要求也大幅提升,公告机构是独立于进行符合性评估活动的产品制造商的第三方机构,要求长期配备具有相关资格证书/资质的产品审查人员、质量管理体系审核人员等,且不能采取外包机制聘用。
对公告机构(NB)的高要求,也让目前获批的能够进行新规公告的机构数量大幅降低。根据欧盟官网信息查询可以看到,过去通过MDD授权的公告机构总计有51家。
然而,自今年5月26日起,这些通过MDD指定的公告机构已不能再根据其指令颁发新证书,而只能对之前颁发的有效证书进行监督过渡。据统计,目前通过欧盟新规MDR授权的公共机构仅有20家。

 分享
分享
图片新闻








技术文库
最新活动更多
-
4月23日立即报名>> 【在线会议】研华嵌入式核心优势,以Edge AI驱动机器视觉升级
-
4月25日立即报名>> 【线下论坛】新唐科技2025新品发布会
-
7.30-8.1火热报名中>> 全数会2025(第六届)机器人及智能工厂展
-
7月30-31日报名参会>>> 全数会2025中国激光产业高质量发展峰会
-
精彩回顾立即查看>> OFweek 2025(第十四届)中国机器人产业大会
-
精彩回顾立即查看>> 【在线会议】从直流到高频,材料电特性参数的全面表征与测量



发表评论
请输入评论内容...
请输入评论/评论长度6~500个字
暂无评论
暂无评论